Overview
· Common anatomical sites: sacrococcygeal region (≈50%), clivus (≈35%), and mobile spine (≈15%).
· Typically affects middle-aged to elderly adults, with a male-to-female ratio of about 2:1.
· It grows slowly but is locally aggressive and infiltrates surrounding bone and soft tissues.
· Distant metastasis occurs in 20–40% of cases, most often to lungs, liver, and bone.
· Histologically characterized by “physaliphorous cells” containing vacuolated cytoplasm.
· WHO 2020 classification identifies three subtypes: conventional, chondroid, and dedifferentiated.
· Dedifferentiated subtype has the worst prognosis and highest metastatic potential.
· Immunohistochemistry is positive for cytokeratin, EMA, S100, and brachyury (a key diagnostic marker).
Clinical Presentation
· Onset is insidious, with symptoms developing over months or years.
· Pain is the most common presenting complaint, often dull, midline, and progressive.
· Neurologic deficits arise from compression of adjacent neural structures.
· Sacral lesions may cause bowel or bladder dysfunction, saddle anesthesia, or constipation.
· Clival lesions may present with diplopia, headache, dysphagia, or cranial nerve palsy (VI, IX, X).
· Cervical or thoracolumbar spine chordomas may lead to radiculopathy or myelopathy.
· A palpable midline mass may be seen in advanced sacral disease.
Imaging
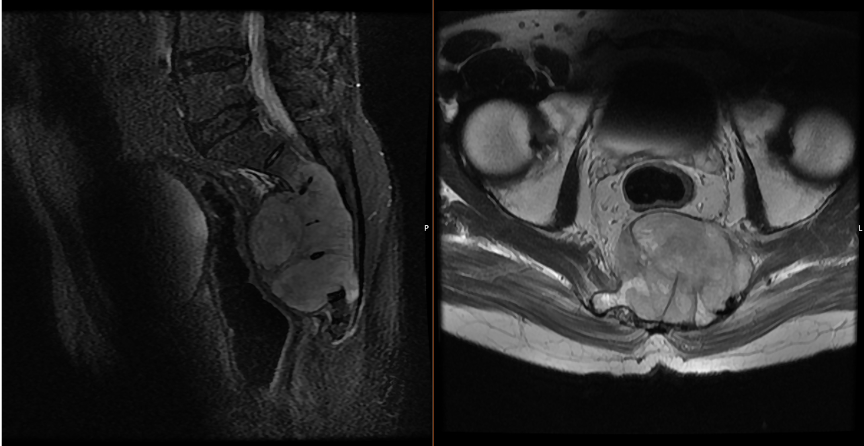
· X-ray: Lytic, destructive midline lesion with cortical thinning or erosion.
· CT scan: Reveals bone destruction with lobulated soft-tissue mass; may show internal calcifications or sequestra.
· MRI: High T2 signal intensity with a lobulated, septated appearance; low-to-intermediate T1 signal.
· Often demonstrates “soap-bubble” or “honeycomb” pattern with enhancement after gadolinium contrast.
· Helps define the tumor’s relation to dura, neural foramina, and vascular structures.
· Diffusion-weighted imaging and fat-suppressed sequences are useful for assessing local invasion.
· PET-CT may assist in detecting recurrence or distant metastases.
Treatment
· Surgical resection with wide or en bloc margins remains the gold standard.
· Aim is gross total resection with negative margins while preserving neurologic function.
· When complete resection is impossible, subtotal resection followed by radiotherapy is recommended.
· Proton-beam or carbon-ion radiotherapy provides superior local control compared to conventional photon therapy.
· High-dose radiation (>70 Gy RBE) improves recurrence-free survival.
· Chemotherapy is largely ineffective except for dedifferentiated subtypes, where doxorubicin-based regimens may be used.
· Immunotherapy and targeted therapy (e.g., PDGFR, EGFR, or mTOR inhibitors) are under investigation.
· Recurrence management involves repeat surgery or stereotactic radiotherapy when feasible.
· Multidisciplinary planning is essential due to complex anatomy and risk of recurrence.
Surgical Indications
· Symptomatic or progressive lesion causing pain or neurological compromise.
· Tumors that are technically resectable with potential for clear margins.
· Recurrent disease amenable to repeat resection.
· Failure of prior radiation or decompression surgery.
· Prevention of impending neurological or visceral dysfunction in advanced sacral cases.
Prognosis
· Despite slow growth, local recurrence is common (50% within 5 years).
· 5-year overall survival: 60%; 10-year survival 25%.
· Favorable prognostic factors: complete resection, chondroid subtype, younger age.
· Poor prognosis associated with dedifferentiation, incomplete resection, or metastasis.
· Median recurrence-free survival ranges from 2 to 5 years after initial treatment.
· Lifelong follow-up with MRI every 6–12 months is recommended due to late recurrences.
Differential Diagnosis
· Chondrosarcoma
· Giant Cell Tumor
· Metastatic carcinoma
· Plasmacytoma or lymphoma:
· Benign notochordal cell tumor:
References
1. Walcott BP, et al. Chordoma: Current concepts, management, and future directions. Lancet Oncol. 2012.
2. Barber SM, Sadrameli SS, Lee JJ, Fridley JS, Teh BS, Oyelese AA, Telfeian AE, Gokaslan ZL. Chordoma-Current Understanding and Modern Treatment Paradigms. J Clin Med. 2021 Mar 4;10(5):1054. doi: 10.3390/jcm10051054. PMID: 33806339; PMCID: PMC7961966.
3. WHO Classification of Tumours of Soft Tissue and Bone, 5th Edition. IARC, 2020.
4. Gronchi A, Miah AB, Dei Tos AP, et al. Soft tissue and visceral sarcomas: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up☆. Ann Oncol. 2021;32(11):1348-1365. doi:10.1016/j.annonc.2021.07.006
5.Boriani S, Bandiera S, Biagini R, et al. Chordoma of the mobile spine: fifty years of experience. Spine (Phila Pa 1976). 2006;31(4):493-503. doi:10.1097/01.brs.0000200038.30869.27.
6. Iannalfi A, D'Ippolito E, Riva G, et al. Proton and carbon ion radiotherapy in skull base chordomas: a prospective study based on a dual particle and a patient-customized treatment strategy. Neuro Oncol. 2020;22(9):1348-1358. doi:10.1093/neuonc/noaa067
7. Jahangiri A, Jian B, Miller L, El-Sayed IH, Aghi MK. Skull base chordomas: clinical features, prognostic factors, and therapeutics. Neurosurg Clin N Am. 2013;24(1):79-88. doi:10.1016/j.nec.2012.08.007
8. Stacchiotti S, Sommer J; Chordoma Global Consensus Group. Building a global consensus approach to chordoma: a position paper from the medical and patient community. Lancet Oncol. 2015;16(2):e71-e83. doi:10.1016/S1470-2045(14)71190-8